|
|
|
|
The field of Structure-Based Drug Design (SBDD) utilizes high-resolution three-dimensional models of protein structure to identify pharmaceutically active compounds. There are two major components of any SBDD algorithm, efficient algorithms for search and accurate models for scoring. Algorithms for SBDD are complicated by the enormity of the search space and the ruggedness of any somewhat reasonable scoring function. An exhaustive brute force search for most protein systems would take time longer than the age of the universe. We obviously can't wait that long! A number of clever search algorithms have helped reduce this time; however, many of these savings in time have come at the cost of reduced preservation of the underlying chemistry. Our lab is currently interested in developing biophysically accurate yet efficient algorithms for molecular modeling.
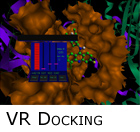 We have been in the field of SBDD for over ten years. In 1996, we helped design an early (perhaps the first) virtual reality based molecular modeling application (figure right). We developed a parallel flexible-ligand Monte Carlo based search algorithm and tied this simulation to an immersive three-dimensional workspace. LCD-shutter based eyewear, motion tracking sensors, and a novel high degree-of-freedom input device completed the immersive experience -- the user was able to fly through the protein active site and guide the simulation through a floating control panel (shown in figure). We have been in the field of SBDD for over ten years. In 1996, we helped design an early (perhaps the first) virtual reality based molecular modeling application (figure right). We developed a parallel flexible-ligand Monte Carlo based search algorithm and tied this simulation to an immersive three-dimensional workspace. LCD-shutter based eyewear, motion tracking sensors, and a novel high degree-of-freedom input device completed the immersive experience -- the user was able to fly through the protein active site and guide the simulation through a floating control panel (shown in figure).
 A few years later, in 1999, we developed one of the first ensemble-based models of molecular flexibility for protein-ligand docking. Our approach utilized multiple protein structure 'snapshots' derived from NMR experiments. A simple aggregate scoring function was used to combine the individual scores of each ligand against each of the structural snapshots. Over the next few years, we identified a number of lead compounds capable of disrupting the binding of Core-Binding Factor alpha to Core-Binding Factor beta. Several years later, in 2007, derivatives of our lead compounds were found to experimentally inhibit CBF dimerization (figure left) and are now being pushed through the drug development pipeline. A few years later, in 1999, we developed one of the first ensemble-based models of molecular flexibility for protein-ligand docking. Our approach utilized multiple protein structure 'snapshots' derived from NMR experiments. A simple aggregate scoring function was used to combine the individual scores of each ligand against each of the structural snapshots. Over the next few years, we identified a number of lead compounds capable of disrupting the binding of Core-Binding Factor alpha to Core-Binding Factor beta. Several years later, in 2007, derivatives of our lead compounds were found to experimentally inhibit CBF dimerization (figure left) and are now being pushed through the drug development pipeline.
We are interested in several lines of SBDD research.
 Improving Binding Mode Prediction: We presented a step toward improving protein-ligand binding mode prediction for a set of ligands known to interact with a common protein. Our algorithm first performs traditional protein-ligand docking for each known binder. The ranked lists of candidate binding modes are then evaluated to identify a set of poses maximally self-consistent with respect to a pharmacophoric map generated from the same poses. We have extensively demonstrated the application of the algorithm to four protein systems (thrombin, cyclin-dependent kinase 2, dihydrofolate reductase, and HIV-1 protease) and attained predictions with an average RMSD below 2.5 A for all tested systems. Improving Binding Mode Prediction: We presented a step toward improving protein-ligand binding mode prediction for a set of ligands known to interact with a common protein. Our algorithm first performs traditional protein-ligand docking for each known binder. The ranked lists of candidate binding modes are then evaluated to identify a set of poses maximally self-consistent with respect to a pharmacophoric map generated from the same poses. We have extensively demonstrated the application of the algorithm to four protein systems (thrombin, cyclin-dependent kinase 2, dihydrofolate reductase, and HIV-1 protease) and attained predictions with an average RMSD below 2.5 A for all tested systems. - Improved Models of Molecular Flexibility and Protein-Ligand Interactions: In support of our ensemble-based K* scoring function for protein design, we developed a family of branch-and-bound conformational search algorithms, extensions to the Dead-End Elimination (DEE) algorithm for side-chain positioning, and epsilon approximation algorithms for computing partition functions. We are interested in translating these ideas to SBDD.
- The Revitalization of de novo SBDD: Along with virtual screening, de novo SBDD once held significant promise. Difficulty in synthesizing identified molecules along with questions of their chemical stability prevented do novo SBDD from taking off. We are interested in integrating our work in Chemical Synthesis Planning (CSP) for lead optimization.
- Optimization of High Throughput Screening (HTS) Ligand Libraries: Although HTS libraries may contain over a million compounds, they are likely to inadequately sample small molecule space. We are investigating the possibility of tuning the ligand library to the protein target before screening begins. The goal is to maximize the chance of obtaining a hit in an HTS screen.
 Seeking Collaborations: Having recently moved to UofT, we are now actively seeking collaborators in structural biology, clinical medicine, and the pharmaceutical industry. We are currently in preliminary discussions with several potential groups. If you're interested please drop us a line. Seeking Collaborations: Having recently moved to UofT, we are now actively seeking collaborators in structural biology, clinical medicine, and the pharmaceutical industry. We are currently in preliminary discussions with several potential groups. If you're interested please drop us a line.

|
|
|
|
|
|
|
|