 |
|
|
|
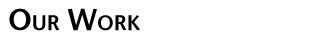
 In the next phase of our protein redesign work, we are interested in improving both the accuracy and speed of current methods and in extending the domains into which algorithms for protein redesign might be applied. For example, our collaborator Professor Kevin Truong is an expert in the design of fluorescent biosensors. Fluorescent biosensors measure the energy transfer between a donor and an acceptor fluorophore (figure right). This phenomenon is referred to as FRET (Fluoresence Resonance Energy Transfer).
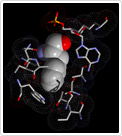
In the next phase of our protein redesign work, we are interested in improving both the accuracy and speed of current methods and in extending the domains into which algorithms for protein redesign might be applied. For example, our collaborator Professor Kevin Truong is an expert in the design of fluorescent biosensors. Fluorescent biosensors measure the energy transfer between a donor and an acceptor fluorophore (figure right). This phenomenon is referred to as FRET (Fluoresence Resonance Energy Transfer).  By attaching the donor and acceptor to a third protein (one capable of binding the desired target ligand), it becomes possible to detect changes in the concentration of a target molecule. FRET efficiency is a function of the relative positioning of the donor and acceptor fluorophores. Using protein redesign to optimize the relative positions of the donor and acceptor fluorophores we are exploring the possibility of creating a suite of generic fluorescent biosensors and biosensor templates. Biologists would be able to request on-demand redesigns of specific sensors. These biosensors would be useful both in molecular biology labs where researchers measure changes in concentration of various electrolytes and metabolites as well as in the clinic where a new generation of fluorescent bio-sentinels could monitor for changes in patient status. These projects pose several difficult algorithmic challenges, including scalability, linker design, and scoring of FRET efficiency. By attaching the donor and acceptor to a third protein (one capable of binding the desired target ligand), it becomes possible to detect changes in the concentration of a target molecule. FRET efficiency is a function of the relative positioning of the donor and acceptor fluorophores. Using protein redesign to optimize the relative positions of the donor and acceptor fluorophores we are exploring the possibility of creating a suite of generic fluorescent biosensors and biosensor templates. Biologists would be able to request on-demand redesigns of specific sensors. These biosensors would be useful both in molecular biology labs where researchers measure changes in concentration of various electrolytes and metabolites as well as in the clinic where a new generation of fluorescent bio-sentinels could monitor for changes in patient status. These projects pose several difficult algorithmic challenges, including scalability, linker design, and scoring of FRET efficiency.
In a more general context, the methods we design for scoring protein-ligand interactions in protein redesign can be applied to modeling other protein-ligand interactions. Our algorithms for efficiently computing ensemble-based measures of protein-ligand affinity will be adopted for use in our Structure-Based Drug Design project.
The following is a bullet-point summary of our previous work on protein redesign. For additional details please see our protein redesign publications.
|
|
|
|
|
|
|
|

 Realization of novel molecular function requires the ability to alter molecular complex formation. Enzymatic function can be altered by changing enzyme-substrate interactions via modification of an enzyme’s active site. A redesigned enzyme may either perform a novel reaction on its native substrates or its native reaction on novel substrates. A number of computational approaches have been developed to address the combinatorial nature of the protein redesign problem. These approaches typically search for the global minimum energy conformation among an exponential number of protein conformations.
Realization of novel molecular function requires the ability to alter molecular complex formation. Enzymatic function can be altered by changing enzyme-substrate interactions via modification of an enzyme’s active site. A redesigned enzyme may either perform a novel reaction on its native substrates or its native reaction on novel substrates. A number of computational approaches have been developed to address the combinatorial nature of the protein redesign problem. These approaches typically search for the global minimum energy conformation among an exponential number of protein conformations.  Developed Restricted Dead-End Elimination (rDEE), a method for efficient search given the constraint of a bounded number of residue mutations.
Developed Restricted Dead-End Elimination (rDEE), a method for efficient search given the constraint of a bounded number of residue mutations.


