FullSignalRanker
FullSignalRanker: Probabilistic Inference on Multiple Normalized Signal Profiles
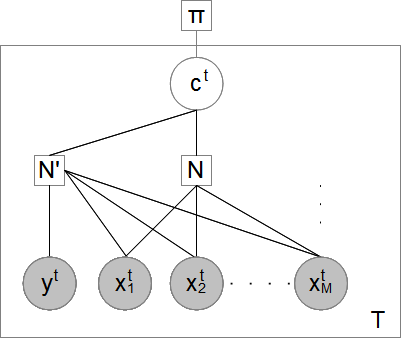
Overview
With the prevalence of chromatin immunoprecipitation (ChIP) with sequencing (ChIP-Seq) technology, massive ChIP-Seq data has been accumulated. The ChIP-Seq technology measures the genome-wide occupancy of DNA-binding proteins in vivo. It is well-known that different combinations of DNA-binding protein occupancies may result in a gene being regulated in different tissues or at different developmental stages. To fully understand a gene's function, it is essential to develop probabilistic models on multiple ChIP-Seq profiles for deciphering the combinatorial gene transcription. To this end, we propose a method (FullSignalRanker) for regression tasks on ChIP-Seq data. The proposed method is compared with other existing methods on ENCODE ChIP-Seq datasets, demonstrating its regression and classification ability. The results suggest that FullSignalRanker is the best-performing method for recovering the signal ranks on the promoter and enhancer regions. In addition, FullSignalRanker is also the best-performing method for peak sequence classification. We envision that FullSignalRanker will become important in the era of next generation sequencing.
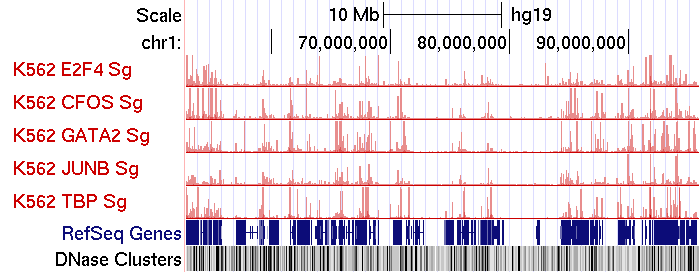
(UCSC Genome Browser ScreenShot on ENCODE ChIP-Seq data)
Downloads
win64 |
linux64 |
|
| MCR (Matlab Compiler Runtime) | ||
| FullSignalRanker Executables and Demo Dataset (zipped) |
Command Usage
FullSignalRanker inFilePath testingFilePath [outModelPath] [numOfCombinations] [threshold] [maxIterations] [replicates]
| Input Arguments: | |
| inFilePath | The input training profile signal file path (example: inData.csv) |
| testingFilePath | The input testing profile signal file path (example: testingData.csv) |
| outModelPath | The output FullSignalRanker model file path (default: FSRmodel.mat) |
| numOfCombinations | Number of Clusters (default: 2) |
| threshold | The tolerance used for testing convergence (default: 0.0001) |
| maxIterations | Maximal number of EM iterations (default: 100) |
| replicates | Number of replicates (default: 10) |
| Output Files: | |
| A FullSignalRanker model file as specified in the input argument "outModelPath" (default: FSRmodel.mat) | |
| Regression result image with the input argument "outModelPath" as the filename prefix. | |
| Microsoft Windows 64-bits examples: | |
| C:\> FullSignalRanker <argument_list> | |
| C:\> FullSignalRanker inData.csv testingData.csv | |
| C:\> FullSignalRanker inData.csv testingData.csv simpleModel.mat | |
| C:\> FullSignalRanker inData.csv testingData.csv fancyModel.mat 10 0.00001 1000 100 | |
| Linux 64-bits examples: | |
| >./run_FullSignalRanker.sh <mcr_directory> <argument_list> | |
| >./run_FullSignalRanker.sh /mathworks/home/application/v80 inData.csv testingData.csv | |
| >./run_FullSignalRanker.sh /mathworks/home/application/v80 inData.csv testingData.csv simpleModel.mat | |
| >./run_FullSignalRanker.sh /mathworks/home/application/v80 inData.csv testingData.csv fancyModel.mat 10 0.00001 1000 100 | |
FAQ
What is MCR ?
MCR is Matlab Compiler Runtime. If your machine does not have Matlab, you need to install MCR to execute FullSignalRanker. MCR can be downloaded from the internet easily. In particular, we advise you to download the same version indicated in the "Downloads" section.
Is there any demo ?
By default, a small pair of training and testing dataset (inData.csv and testingData.csv) is zipped with the FullSignalRanker executables in the "Downloads" section. Once downloaded, you can simply change your current directory to it and type "FullSignalRanker inData.csv testingData.csv" to run a FullSignalRanker demo on the testing dataset (which has 5 predictor profiles and 1 response profile (last column) under 3 clusters). After the run, you will see the MATLAB FullSignalRanker model file (default: FSRmodel.mat) and the regression result image file as follow:
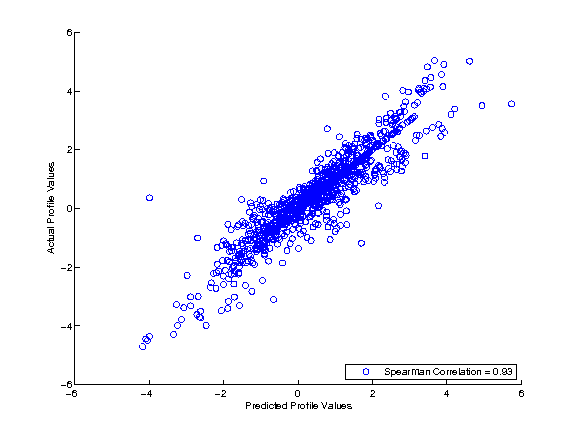
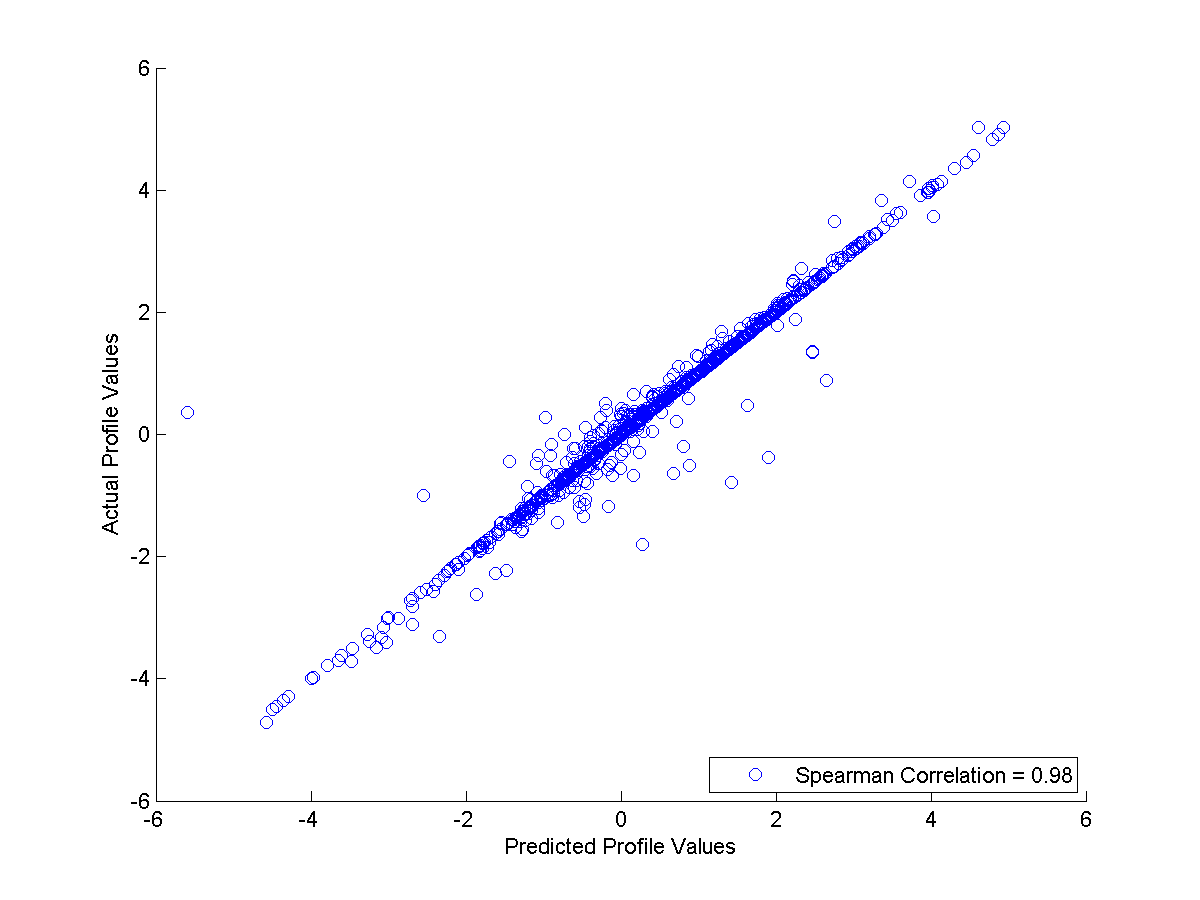
Since the data is generated by 3 clusters, the default setting is not suitable actually. Therefore, you can try it again with the correct parameter setting by typing "FullSignalRanker inData.csv testingData.csv FSRmodelC3.mat 3". After the run, you will get a better regression result image like this:
More data ?
Public ChIP-Seq data can be accessed through ENCODE consortium and Gene Expression Omnibus (GEO). Details of Wiggler can be found here.
More questions ?
Please contact Ka-Chun Wong