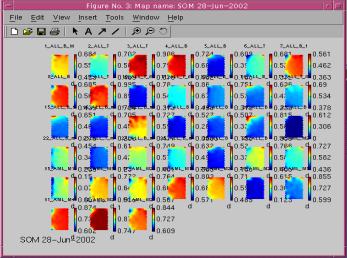
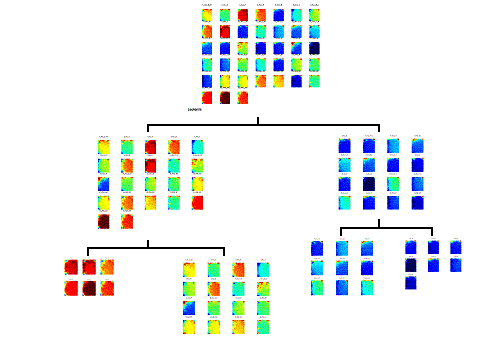
|
Introduction
|
|
First time users, please follow following analysis steps: Load data ŕ Plot data ŕ Apply normalizationŕ Plot data, if required normalize again, or select another normalization, and plot data ŕ Select “Specimens” or “Gene” tab to decide which space you are going to cluster ŕ Decide SOM topology, or press topology tab for software to suggest topology. ŕ Compute SOM, if the component planes are not visually distinguishable and are homogenous, select some different topology or do other normalization, and repeat all the above steps.ŕ Press “Partitive k-means” tab for partitive clustering. ŕ Finally hit “BTSVQ” tab to generate clustering results. All the steps can be repeated in any order, after completing first round, except BTSVQ. To generate another set of results “Partitive k-means” tab should pressed before “BTSVQ”.
Figure (1)
|
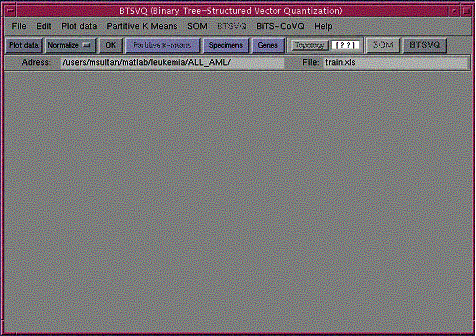
|
2. Loading Data
|
|
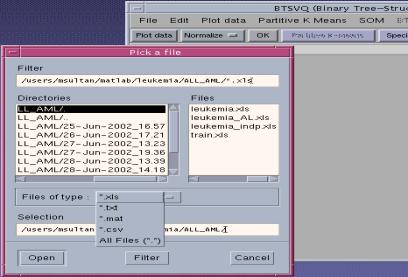
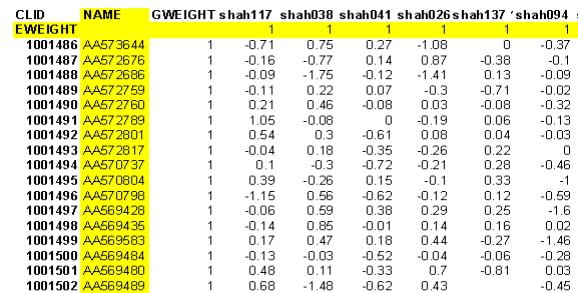
Following file formats are supported. 2.1 ASCII text filesA typical Microarray text file is shown in the Figure (3), First row and first column are taken as the specimen labels and gene labels respectively. The file with more than one row or columns of the descriptors of gene labels or specimen labels can also be loaded. You will be asked of the total number of columns in the text file. Also you will be prompted for the number of gene label columns and specimen label rows in the file, and only first row and first columns will be taken as Specimen label and Gene ID, the rest of the header lines will be discarded, and the data will be loaded beyond that range, as shown in figure (3), the yellow lines will not be included in the analysis.
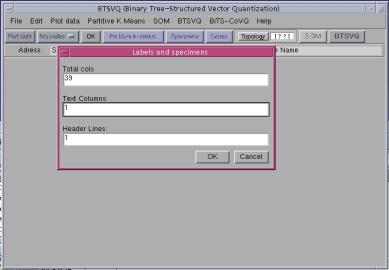
To load ASCII text files, total number of columns in the file and number of text columns and header lines should be specified.
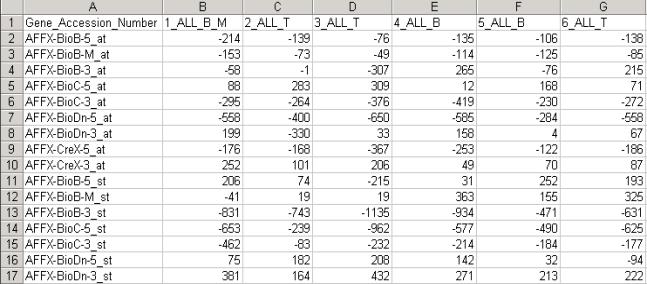
Figure(4) 2.2 Microsoft Excel filesThe preferred format of the excel files is shown in Figure (5). If there are more than one text columns or rows for gene ID’s and specimen labels, only first row and column will be considered in the analysis and the rest would be discarded. Also note that specimen and gene labels should be text or Alphanumeric, numeric entries should be changed to alphanumeric. Column A, and Row 1 should have text in it otherwise an XLS read error will appear.
Figure(5) 2.3 Comma Separated (CSV) files.Coma separated files are also loaded like text files, by specifying total number of columns in the file and number of text columns and header lines. 2.4 Matlab .mat format filesMat files with following variables present can also be loaded
|